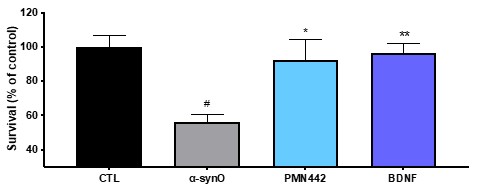
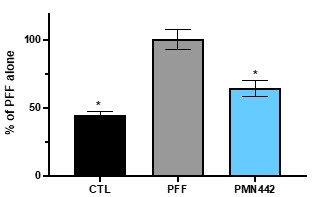
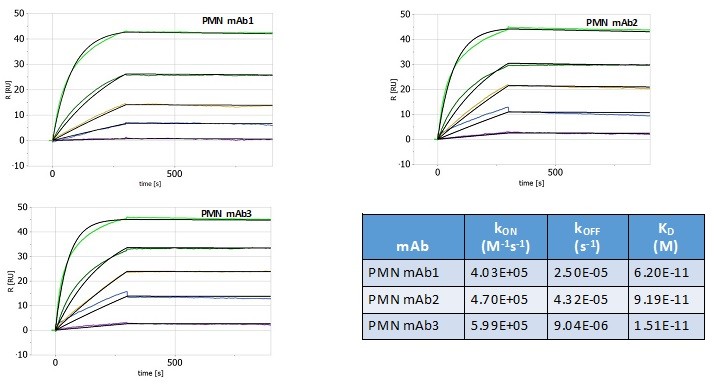
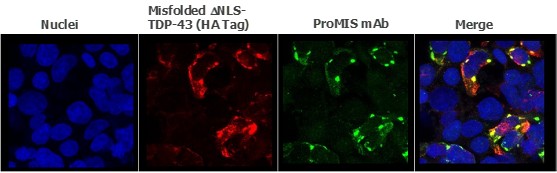
Pursuant to its charter, the Audit Committee is charged with oversight over related party transactions entered into by the Company and conducts an appropriate review of all related party transactions for potential conflict of interest situations on an ongoing basis. We do not have a separate related person transaction policy.
Company Transactions with Related Parties
The Company has entered into related party transactions as follows:
Neil Cashman. In April 2016, the Company entered into a three-year, collaborative research agreement (“CRA”) with UBC and the Vancouver Coastal Health Authority in the amount of C$787,500, with Dr. Cashman as principal investigator at UBC. In March 2018, the CRA was amended and funding was increased to C$892,500 over three years. In July 2018, the total funding commitment to UBC increased to C$1,130,000 over the period of the agreement. In February 2019, the CRA was amended, and funding was increased to C$2,130,000 for an additional two-year period. In September 2019, the CRA was amended, and funding was increased to C$2,630,000 for an additional one- year period. In November 2021, the CRA was amended for an additional grant of C$800,000 effective January 1, 2022, for the 2022 calendar year for total funding of C$3,430,000. The Company incurred costs of $557,665 and $393,341 for the years ended December 31, 2022 and 2021, respectively, which are included in research and development expenses in the accompanying consolidated statements of operations and comprehensive loss. Dr. Cashman is Professor Emeritus with UBC. Please see “Business” for more information regarding the Company’s relationship with UBC. For additional information regarding this agreement and the amounts paid for Mr. Cashman’s services, see Item 11 - “Executive Compensation.”
Eugene Williams. The Chairman and CEO services provided by Mr. Williams to the Company were historically provided pursuant to a consulting agreement entered into between the Company and Virtua, LLC dated June 29, 2015. For additional information on such consulting agreement, please see Item 11 - “Executive Compensation.”
Daniel Geffken. The CFO services provided by Mr. Geffken are provided pursuant to a consulting agreement entered into between the Company and Danforth Advisors, LLC dated October 17, 2016, and as amended from time-to-time (the “Danforth Consulting Agreement”). Under the Danforth Consulting Agreement, Mr. Geffken agreed to provide the Company the customary services of a CFO at an hourly rate of $325 for a one year term. On March 27, 2017, the Danforth Consulting Agreement was amended to provide for services based on a $5,000 monthly retainer, subject to a 4% annual increase, plus expenses. The Danforth Consulting Agreement was subsequently amended on December 12, 2017 to extend the term for an additional year and on August 31, 2018 to extend the term for an additional year. The Danforth Consulting Agreement was further amended on November 10, 2021 to extend the term of the consulting agreement through October 29, 2024 and to set Mr. Geffken’s compensation at a fixed monthly fee of $15,000. For the years ended December 31, 2022 and 2021, the Company paid $365,247 and $322,639, respectively, to Danforth Advisors, LLC for services provided pursuant to a consulting agreement. The Danforth Consulting Agreement provides for an extension of terms by the mutual agreement of the parties and that either party may terminate the agreement upon sixty days prior written notice to the other, or 30 days in the case of termination for cause. Additionally, on March 1, 2017 and November 12, 2021, the Company granted 8,333 and 8,333 share options, respectively, to Danforth Advisors, LLC pursuant to the same agreement, which will vest as follows: 25% vested immediately upon the grant of options and the balance will vest in equal installments over 36 months.
Larry Altstiel. On April 1, 2022, in connection with appointment as part-time CMO of the Company, the Company entered into a consulting agreement with Dr. Altstiel (the “Altstiel Consulting Agreement”). Pursuant to the Altstiel Consulting Agreement, Dr. Altstiel serves as CMO of the Company until April 1, 2023 (unless terminated earlier) and, in exchange for such consulting services, the Company pays Dr. Altstiel a month fee of $19,000 plus reimbursement of reasonable, out-of-pocket expenses or disbursements incurred in connection with his performance as CMO. On April 14, 2022, the Company also awarded Dr. Altstiel 30,833 stock options pursuant to the terms of the Stock Option Plan (the “Altstiel Consulting Options”). The Altstiel Consulting Options will vest in equal monthly portions over 48 months.
Please see “Executive Compensation” for details regarding named executive officer and director compensation.